Computer-Aided Drug Design CADD: Types, Uses, Examples, Softwares
Table Of Content
- Unraveling the Genomic Landscape: An In-Depth Look at ChIP-seq Library Preparation and Data Analysis
- 4 Docking-based VS
- What happens to the research data of drug molecules that stop at pre-clinical stage?
- Ligand-Based Drug Design
- 2 Site Identification by Ligand Competitive Saturation (SILCS)
- Next frontier for drug discovery: SPACE?
- 4. Site identification using SILCS-Hotspots
In this chapter, standard CADD protocols for both SBDD and LBDD will be presented with a special focus on methodologies and targets routinely studied in our laboratory for antibiotic drug discoveries. Structure-based and ligand-based drug design form two branches of the computer-aided drug discovery process which plays a significant role in the design and identification of drug molecules in reduced time and cost. The increase in the number of positive cases and deaths from COVID-19 and the lack of approved drugs and vaccines continue to be a matter of global health concern which necessitates the urgent discovery of drugs for the prevention and cure of the disease.
Unraveling the Genomic Landscape: An In-Depth Look at ChIP-seq Library Preparation and Data Analysis
The primary objective of CADD is to screen, optimize and evaluate the activity of the compound against the target. It forms the multi-disciplinary approach for both academic and major pharmaceutical companies for better efficacy with no/fewer side effects. To speed up virtual screening of ultra-large chemical libraries, several groups have suggested hybrid iterative approaches, in which results of structure-based docking of a sparse library subset are used to train ML models, which are then used to filter the whole library to further reduce its size.
4 Docking-based VS
In addition to their enormous sizes, these virtual spaces are highly novel and diverse, and have minimal overlap (less than 10%) between each other46. Currently, the largest commercial space, Enamine REAL Space, is an extension to the REAL database that maintains the same synthetic speed, rate and cost guarantees, covering more than 170 reactions and more than 137,000 building blocks (Box 1). Most of these reactions are two-component or three-component, but more four-component or even five-component reactions are being explored, enabling higher-order combinatorics. This space can be easily expanded to 1015 compounds based on available reactions and extended building block sets, for example, 680 million of make on demand (MADE) building blocks47, although synthesis of such compounds involves more steps and is more expensive.
What happens to the research data of drug molecules that stop at pre-clinical stage?
Some of the chemical features used in pharmacophore modeling include hydrogen bond donors, hydrogen bond acceptors, aromatic ring systems, hydrophobic areas, positively charged ionizable groups, and negatively charged ionizable groups [79]. Ligands having different scaffolds but the similar spatial arrangement of key interacting functional moieties can be identified using pharmacophore-based virtual screening. The bioactive conformation of the molecules within the target binding site can be incorporated into the pharmacophore model. Some frequently used programs which allow automatic construction of the pharmacophore model include Catalyst, PHASE, LigandScout, GALAHAD, and PharmMapper (Table 3). A good pharmacophore model also incorporates spatial constraints in regions occupied by inactive molecules and often optimized further to make the model less restrictive. All the pharmacophoric features which are not consistently detected in active molecules are either made optional or removed from the final model [7].
Ligand-Based Drug Design
Computer-aided drug discovery has been around for decades, although the past few years have seen a tectonic shift towards embracing computational technologies in both academia and pharma. This shift is largely defined by the flood of data on ligand properties and binding to therapeutic targets and their 3D structures, abundant computing capacities and the advent of on-demand virtual libraries of drug-like small molecules in their billions. Taking full advantage of these resources requires fast computational methods for effective ligand screening.
Modular synthon-based approaches
Why Is TikTok Parent ByteDance Moving Into Biology, Chemistry And Drug Discovery? - Forbes
Why Is TikTok Parent ByteDance Moving Into Biology, Chemistry And Drug Discovery?.
Posted: Wed, 03 Jan 2024 08:00:00 GMT [source]
The latter is based on easily accessible on-demand or generative virtual chemical spaces, as well as structure-based and AI-based computational tools that streamline each step of the drug discovery process. At a deeper level, the results of accurate physics-based docking (in addition to experimental data, for example, from PDBbind81) can be used to train generalized graph or 3D DL models predicting ligand–receptor affinity. This would help to markedly expand the training dataset and balance positive and negative (suboptimal binding) examples, which is important to avoid the overtraining issues described in ref. 87. Such DL-based 3D scoring functions for predicting molecular binding affinity from a docked protein−ligand complex are being developed and benchmarked, most recently RTCNN98, although their practical utility remains to be demonstrated. Molecular descriptors are numerical values used to represent the structural and physicochemical properties of molecules [53, 54]. The molecular descriptor field is strikingly interdisciplinary and includes a number of different theories [55].

Boxes with solid lines indicate methods used in the CMY-10 study while boxes with dashed lines mark methods not used in the CMY-10 study but in other studies. Double arrows indicate the two techniques can be used interactively in several iterative drug design rounds. Although blind benchmarking and recent prospective screening success stories for the growing number of targets support utility of modern computational tools, there are whole classes of challenging targets, in which existing in silico screening approaches are not expected to fare very well by themselves.
Since then CADD methods have been employed extensively to facilitate the development of novel antibiotics by the computational chemistry community and us for the past five years. This included studies on the mechanism behind antibiotic resistance that may help to guide the design of new antibiotic drugs to overcome such resistance. Al. studied the mechanism of a S222T mutation induced resistance of Deoxy-D-xylulose 5-phosphate reductoisomerase (DXR) to fosmidomycin using molecular dynamics (MD) simulations (5). The MD simulations revealed the structural and energetic basis of the single mutation that induced resistance shedding light on the development of a new antibiotic compounds targeting DXR. Al. recently explored the molecular mechanism of polymyxin E (colistin) resistance in mobile colistin-resistant (mcr-1) bacteria (6). Colistin is the only FDA-approved membrane-active drug to tackle Gram-negative bacteria despite its high toxicity.
This synergy is reflected in the latest 3DR Grand Challenge 4 results for ligand IC50 predictions59, in which the top methods that used a combination of both physics-based and ML scoring outperformed those that did not use ML. Going forward, thorough benchmarking of physics-based, ML and hybrid approaches will be a key focus of a new Critical Assessment of Computational Hit-finding Experiments (CACHE), which will assess five specific scenarios relevant to practical hit and lead discovery and optimization97. The modular nature of on-demand virtual libraries supports further growth by the addition of reactions and building blocks.
POLARISqb And Scientist.com Partner to Offer Online Access to Quantum-Aided Drug Design - The Quantum Insider
POLARISqb And Scientist.com Partner to Offer Online Access to Quantum-Aided Drug Design.
Posted: Mon, 08 Jan 2024 08:00:00 GMT [source]
Notably, CADD methods are evolving with researchers continually updating and implementing new CADD techniques with higher levels of accuracy and speed (24–26). In this chapter, we will present commonly used CADD approaches, including those used in our lab for the design of next-generation antibiotics. A pharmacophore model elucidates the spatial arrangement of chemical features in ligands that are required for interaction with the target receptor [78].
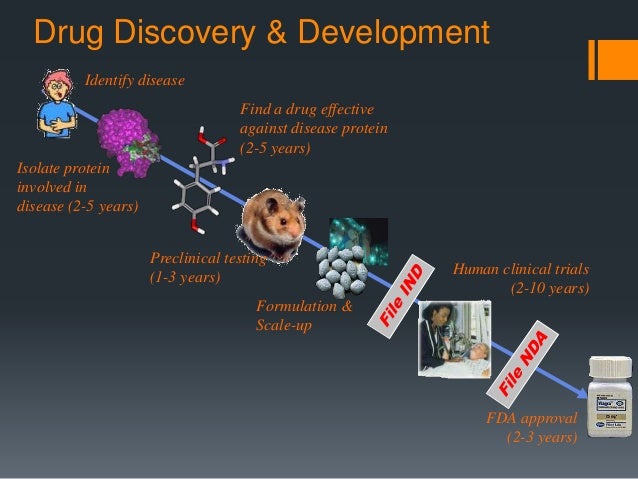
Some of the common methods employed in SBDD include structure-based virtual screening (SBVS), molecular docking, and molecular dynamics (MD) simulations. These methods find numerous applications such as assessment of binding energetics, protein-ligand interactions, and conformational changes in the receptor upon binding with a ligand [20]. Being used by many pharmaceutical industries and medicinal chemists, SBDD as a computational technique has greatly helped in the discovery of several drugs available in the market. The basic steps involved in SBDD consist of the preparation of target structure, identification of the ligand binding site, compound library preparation, molecular docking and scoring functions, molecular dynamic simulation, and binding free energy calculation (Figure 1). The recent outbreak of the deadly coronavirus disease 19 (COVID-19) pandemic poses serious health concerns around the world.


Comments
Post a Comment